| Introduction Receptor-mediated gene transfer has been shown to be effective for delivering genes into tissues by specific cellular receptors. Using this method, DNA is usually conjugated to a carrier molecule with a ligand domain specific for a cell surface receptor and a DNA-binding domain, a poly-L-lysine (PL) peptide. This DNA–ligand complex is internalized by targeted cells when the ligand binds to its respective cell-surface receptor. Such receptor-mediated gene transfer has been accomplished using a variety of receptors by conjugating DNA to ligands such as asialo-orosomucoid,1,2 transferrin,3,4,5 lectins,6 folate,7 lung surfactant protein,8 insulin,9 and monoclonal antibodies.10,11 Receptor-mediated gene transfer has some advantages over the other methods of in vivo gene transfer. Compared with attenuated viral vectors, it shares tissue-specificity, but receptor-mediated gene transfer minimizes the use of viral gene elements, obviating the concerns regarding genomic integration. Further, it lessens concerns with the proinflammatory properties often associated with viral vectors.12,13,14 The DNA–ligand complex is believed to be internalized by receptor-dependent endocytosis rendering transfection to be minimally nontoxic. The conjugate carrier can be designed for cell-specific targeting by selecting the appropriate receptor ligand. Past studies have shown that oral and intranasal immunizations with soluble proteins and in conjunction with mucosal adjuvants are able to induce mucosal immune responses evident by elevated mucosal IgA antibodies. However, soluble protein immunization is limited in its requirement of multiple deliveries due to inefficient antigen uptake by mucosal inductive tissues which often may require deleterious adjuvants. To facilitate antigen uptake, attenuated live vector systems such as Salmonella can target mucosal inductive tissues via interaction with Peyer’s patch M cells.15 M cells or membrane-like epithelium cells compose the thin epithelial layer above the Peyer’s patch dome. These M cells are responsible for sampling the gut lumen for antigens, thus, mediating antigen presentation to gut mucosal inductive tissues. Subsequently, presentation of antigen can stimulate mucosal B and T cells. The limitation of live vector systems is that these attenuated vectors can often cause mild disease and may be problematic for immunocompromised subjects. To circumvent such potential problems, it would be advantageous to devise a vaccine vehicle that exhibits the targeting properties of live vector delivery systems, but lacks the risk of producing mild disease. Reovirus is an enteric pathogen and infects the host following attachment to intestinal Peyer’s patch M cells.16,17 Thus, as with other enteric pathogens, reovirus exploits M cells as a means to gain entry into the host. Mediating reovirus attachment is the adhesin, protein 1, which is expressed and located at the 12 vertices of the viral icosahedron representing the tips of the spikes.16 The protein 1 is a 45 kDa protein that polymerizes via its N-terminus17 to form a tetramer when isolated from reovirus-infected cells or purified as a recombinant protein from E. coli,18 although it is believed to exist as a trimer when attached to the viral icosahedron.19 Considering the ability of protein 1 to bind M cells, we questioned whether this protein could be used to mediate gene transfer, and ultimately, aid in the delivery of DNA for mucosal immunization. Herein we report the development of a DNA delivery vehicle based on reovirus protein 1. The objective for these studies is to demonstrate that receptor-mediated DNA transfection can be effectively performed using recombinant fusion protein 1. The recombinant fusion protein 1 was modified by covalently coupling it to PL. The resulting protein 1-PL conjugate readily forms a complex with DNA, presumably through electrostatic interactions. With this conjugate–DNA complex, we can successfully transfect rodent and human cells. In addition, ex vivo binding studies demonstrated the binding of recombinant fusion protein 1 to nasal-associated lymphoid tissues (NALT) suggesting that protein 1-based vehicle can be applied for genetic vaccination of mucosal tissues. ResultsThe cell binding capacity of recombinant protein 1 Recombinant reovirus protein 1 has been shown to bind to mouse L cells.20 To test whether our recombinant protein 1 as a maltose binding protein (MBP) fusion derived from an E. coli expression system retained its ability to bind L cells, immunofluorescent staining was performed using a biotinylated monoclonal antibody to reovirus protein 1 and streptavidin-phycoerythrin (SA-PE), and analyzed by flow cytometry. As depicted, protein 1 was able to bind mouse L cells, rat fetal lung RFL-6 fibroblasts, and human Caco-2 intestinal epithelial cells (Figure 1a, b and c). In the absence of protein 1, no immunofluorescence was detected. Thus, the recombinant protein 1 maintains its cell-binding ability of wild-type reovirus protein 1. In addition, this cell-binding capacity was only slightly affected when protein 1 was covalently linked to PL as shown by the reduced immunofluorescence (Figure 1d) when compared with the unmodified protein 1 (Figure 1a). Quantitation of transfection efficiency After transfection with 1-PL-pCMV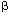 -Gal, expression of -gal in mouse L cells was determined. Transfection with protein 1-PL-pCMV-Gal complexes produced a statistically significant higher level of -gal expression (Figure 2A) than that obtained with PL-pCMV-Gal (Figure 2B) in both L and RFL-6 cells. Preliminary staining showed that Caco-2 cells exhibited endogenous -gal activity, and subsequently, were not evaluated (data not shown). Transfection efficiencies for L cells (Table 1) and RFL-6 cells (Table 2) were 7.2% and 4.7%, respectively. Chloroquine treatment significantly enhanced protein 1-PL-mediated transfection and resulted in about a 2.2-fold increase in transfection efficiency for both cell lines (Tables 1 and 2). Transfection with PL-pCMV-Gal (lacking protein 1-mediated targeting) was ineffective and levels of -gal expression did not differ from background levels. -Gal, expression of -gal in mouse L cells was determined. Transfection with protein 1-PL-pCMV-Gal complexes produced a statistically significant higher level of -gal expression (Figure 2A) than that obtained with PL-pCMV-Gal (Figure 2B) in both L and RFL-6 cells. Preliminary staining showed that Caco-2 cells exhibited endogenous -gal activity, and subsequently, were not evaluated (data not shown). Transfection efficiencies for L cells (Table 1) and RFL-6 cells (Table 2) were 7.2% and 4.7%, respectively. Chloroquine treatment significantly enhanced protein 1-PL-mediated transfection and resulted in about a 2.2-fold increase in transfection efficiency for both cell lines (Tables 1 and 2). Transfection with PL-pCMV-Gal (lacking protein 1-mediated targeting) was ineffective and levels of -gal expression did not differ from background levels. Protein 1-PL mediated transfer of luciferase reporter gene To determine if protein 1 could mediate transfection with other than pCMV-Gal, mouse L cells were transfected using protein 1-PL-pCMVLuciferase (Luc). L cells were exposed to 2 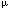 g pCMVLuc complexed with 8 g 1-PL, and 24 h later were assessed for expression of Luc. Luc activity (486940 g pCMVLuc complexed with 8 g 1-PL, and 24 h later were assessed for expression of Luc. Luc activity (486940 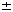 43954) could be detected in several independent experiments (n = 6) as a consequence of protein 1-PL-pCMVLuc transfection (Figure 3). Significant expression of Luc was also achieved with RFL-6 and Caco-2 cells, of which Luc activities were 40684 6633 (n = 6) and 40703 6225 (n = 6), respectively. The transfection did appear to be mediated by protein 1 when compared with the various control transfections. The background activity of Luc is 193 29 (n = 6). Minimal detectable Luc activity could be measured when L cells were transfected with pCMVLuc only, protein 1 associated, but not covalently attached with PL-pCMVLuc, or PL complexed with pCMVLuc without any protein 1 (Figure 3). Thus, optimal transfection required the covalent attachment of protein 1 for optimal cellular transfection. 43954) could be detected in several independent experiments (n = 6) as a consequence of protein 1-PL-pCMVLuc transfection (Figure 3). Significant expression of Luc was also achieved with RFL-6 and Caco-2 cells, of which Luc activities were 40684 6633 (n = 6) and 40703 6225 (n = 6), respectively. The transfection did appear to be mediated by protein 1 when compared with the various control transfections. The background activity of Luc is 193 29 (n = 6). Minimal detectable Luc activity could be measured when L cells were transfected with pCMVLuc only, protein 1 associated, but not covalently attached with PL-pCMVLuc, or PL complexed with pCMVLuc without any protein 1 (Figure 3). Thus, optimal transfection required the covalent attachment of protein 1 for optimal cellular transfection. To show specificity of protein 1-mediated gene transfer, transfection of L cells was performed using protein 1-PL-pCMVLuc in the presence of excess, unconjugated recombinant protein 1 or in the presence of an anti-reovirus polyclonal antibody. If indeed, the cell transfection was receptor-mediated, these molecules should inhibit transfection. As such, in the presence of increasing concentrations of unconjugated protein 1 resulted in the attenuation of gene transfer (Figure 4a) with an IC50 of approximately 75 g/ml for protein 1. Between 100 and 500 g/ml of uncomplexed protein 1 resulted in nearly 100% inhibition of cell transfection. As for transfection of Caco-2 cells, 100 and 500 g/ml of uncomplexed protein 1 caused a respective 89% and 94% inhibition of Luc expression (data not shown). Protein 1-mediated transfection was receptor-specific since bovine serum albumin (BSA) could not inhibit protein 1-PL-mediated gene transfer. The gene expression in the presence of BSA at 0.5 mg/ml was 899000 145720 light units (n = 3) (data not shown). As with the recombinant protein 1, the polyclonal anti-reovirus 3 antibody showed inhibition of the protein 1-PL-pCMVLuc gene transfer to mouse L cells in a dose-dependent fashion (Figure 4b). Maximal inhibition was obtained with 10-fold diluted anti-reovirus 3 antibody resulting in a 98% decrease in Luc gene expression (Figure 4b). Normal rabbit IgG did not inhibit protein 1-PL-mediated gene transfer. Interestingly, dilute rabbit anti-reovirus 3 antibody (1:100) appeared to slightly enhance transfection with this DNA complex that may be due to a prozone effect from antibody and protein 1 concentrations. Collectively, these findings demonstrate that protein 1-PL mediated gene transfer is accomplished via ligand binding to target cells. Chloroquine is a lysosomotropic agent, and it is often used in in vitro DNA transfections. It improves transfection presumably by inhibiting DNA degradation in lysosomes.21 However, while chloroquine is often toxic to cells, lesser doses of DNA are required for successful transfection. To test the enhancement of cell transfection by chloroquine treatment using protein 1-PL-pCMVLuc complexes, 2 g of protein 1-PL and 2 g of DNA were used. A 10- to 20-fold increase in cell transfection was obtained as a result of chloroquine treatment when compared with those cells transfected with protein 1-PL-pCMVLuc only (Figure 5). Chloroquine treatment resulted in 10-fold increases in RFL-6 cells, 12-fold increases in mouse L cells, and 20-fold increases in Caco-2 cell transfection. However, chloroquine greatly reduced Luc expression when the ratio of protein 1-PL to DNA was increased. With chloroquine treatment, light units produced by protein 1-PL ratio at the other ratios showed a dose-dependent reduction in transfection (data not shown). Increased cell death was observed at these higher protein 1-PL to DNA ratios when in the presence of chloroquine. Optimization of Luc gene expression following transfection of L cells with protein 1-PL-pCMVLuc To optimize cell transfection, various ratios of DNA to protein 1-PL were evaluated. L cells were exposed to 2 g pCMVLuc complexed with varying amounts of protein 1-PL. As depicted in Figure 6, the optimal amount of protein 1-PL for maximal transfection for 2 g of pCMVLuc was 8 g protein 1-PL or at a ratio of 1:4. Luc expression was also related to the specific amount of plasmid used (Figure 7). Using the fixed DNA:protein 1-PL ratio of 1:4, transfection of L cells was performed with 2, 4, 6 and 8 g pCMVLuc. Maximal Luc expression was obtained between 4 and 8 g of plasmid. Ex vivo binding of recombinant protein 1 to NALT Reovirus entry into the host is mediated by protein 1 via the Peyer’s patches. Since NALT also have an analogous M cell structure,22,23 we hypothesized that reovirus protein 1 should gain entry into the NALT. To test whether the recombinant fusion protein 1 can bind to murine NALT, an ex vivo binding assay was performed. Recombinant protein 1 was able to bind to the NALT (Figure 8A). Control sections, incubated with SA-horseradish peroxidase (HRP) only, failed to stain. Moreover, the binding of biotinylated fusion protein 1 could be competitively inhibited by 10-fold excess unlabeled fusion protein 1 (Figure 8B). Thus, the data suggest that the recombinant protein 1 can specifically bind to the NALT. DiscussionReceptor-mediated gene transfer features the unique ability to target cells for transfection specified by the conjugated ligand. A significant portion of those studies employing receptor-mediated gene transfer used PL–DNA complexes coupled with specific ligands for cell surface receptors.1,4,5,6,7 Such features enable transfection of diverse cells. In light of these studies, we questioned whether a ligand for mucosal inductive tissues could be used as a targeting molecule to facilitate gene transfer of vaccine candidate molecules. Within this report, the objective for the described studies was to combine the attractive features of DNA and live vector delivery systems to devise a DNA delivery system that can potentially target mucosal inductive tissues. Considering that many infectious agents enter the host via a mucosal surface, it may be more advantageous to include within a delivery system, a ligand that specifically targets mucosal inductive tissues.24,25,26,27 Before pursuing such immunization studies, the described work focused on whether receptor-mediated transfection using the recombinant reovirus protein 1 was feasible. Reovirus protein 1 interacts with surface receptors on a variety of cells including erythrocytes,28,29 lymphocytes,29 Peyer’s patches,30,31 and neural cells.32 It is this interaction that allows reovirus to infect the host presumably via the protein 1–M cell interaction.30,31 M cells facilitate antigen sampling allowing uptake by antigen presenting cells located proximal to M cells in the mucosal inductive tissues such as Peyer’s patch.33 Thus, we hypothesize that it would be feasible to manipulate mucosal immunity by M cell-directed gene transfer using protein 1 as a targeting molecule. Due to its surface receptor for reovirus protein 1’s, mouse L cells have been used to propagate reovirus in vitro and to study reoviral attachment.16,17,19,20,24 Evident from our studies is that the recombinant protein 1 retained its ability to bind L cells as a fusion protein when conjugated in a PL–DNA complex. Thus, the E. coli-expressed recombinant protein retained its binding capacity to L cells implicating that the binding domains of the recombinant protein 1 were functionally intact. Further, its expression as a MBP fusion protein did not interfere with its ability to bind to L cells. This binding to L cells was not mediated via the MBP moiety since MBP alone failed to mediate transfection of L cells to the magnitude that was obtained with the recombinant fusion protein. Rat lung fibroblast, RFL-6, and human intestinal epithelial cells, Caco-2, were found to express the surface receptor for protein 1 evident by protein 1’s ability to bind to both cell lines. To test whether protein 1 in a PL–DNA comlex could mediate transfection, the protein 1-PL–DNA complexes were found to be efficiently endocytosed by mouse L cells, as well as RFL-6 and Caco-2 epithelial cells. Elevated levels of Luc activity were obtained only when protein 1 was covalently attached to this complex. The formation of the complex was important for effective gene transfer because simply associating protein 1 with DNA or PL–DNA failed to achieve effective cellular transfection. Further, this transfection was ligand-dependent since the DNA transfection was inhibited with unconjugated protein 1 or with a polyclonal antibody to reovirus 3 expressing reactive antibodies to protein 1. MBP, BSA and normal rabbit IgG had minimal impact upon protein 1-mediated gene transfer. Although low transfection efficiency was obtained, presumably, chloroquine, an agent that inhibits lysosomal degradation and may facilitate DNA release from the vesicle, has been shown to improve gene transfer and expression as evident in other receptor-mediated gene transfer systems.34,35 In this study, chloroquine also enhanced protein 1-PL-mediated gene transfer more than two-fold when pCMV-Gal, and up to 10- to 20-fold when pCMVLuc was used to assess reporter gene expression. However, chloroquine failed to enhance transfection when the ratio (wt/wt) of protein 1-PL to DNA was increased above one, because chloroquine in combination with increased amounts of PL appeared to be toxic to L cells. This combination effect by PL and chloroquine has been previously demonstrated.21 Thus, our studies clearly demonstrate the feasibility of receptor-mediated gene transfer by recombinant protein 1, and they implicate the potential of protein 1-PL–DNA complexes for their application in mucosal immunization. Recent studies have shown the utility of DNA vaccination for inducing protective immunity in experimental animals exposed to influenza,36,37 herpes simplex virus (HSV),38 HIV-1,39,40 rotavirus,41,42 and Borrelia burgdorgeri infections.43 DNA immunization has a number of attractive features including ease of preparation for encoding desired protective immunogens, co-expression of immunogens, co-expression of adjuvant, eg cytokines, lack of large-scale protein purifications, and ease of delivery. DNA vaccines have also been delivered to mucosal surfaces to achieve mucosal immunity. Specific immunity to HSV glycoprotein B was induced using a plasmid DNA expression vector delivered intranasally together with the mucosal adjuvant, cholera toxin, but failed to provide protective immunity upon challenge with wild-type HSV.44 Perhaps a mode to protect the naked DNA from mucosal barriers may improve immunity. As such, elevated mucosal and serum IgA responses were obtained following oral immunization with microencapsulated DNA.45 Likewise, in a separate study, microencapsulated DNA encoding rotavirus VP6 stimulated humoral immunity and reduced rotaviral shedding following challenge.42 Alternatively, cationic liposomes, which enhance the efficiency of DNA delivery, have been used to enhance DNA immunization by increasing the antibody titers.46,47 However, the toxicity associated with cationic liposomes may limit its application. Collectively, these studies suggest that a mode to circumvent mucosal barriers is needed to allow for improved mucosal transfection, and ultimately, for better mucosal immunity. Targeting of host mucosal inductive tissues may improve such immunity. Our findings showing the ability of the protein 1 to mediate efficient gene transfer and bind to the NALT, implicate that immunity to intranasally delivered DNA as part of protein 1 complex is achievable. Current studies are evaluating such possibilities. This evidence suggests that protein 1-directed gene transfer may be an efficient mode for targeting mucosal inductive tissues. Thus, the protein 1-PL-mediated vaccination system may represent an alternative means for mucosal vaccination. Materials and methodsProduction of recombinant reovirus 1 protein The cloned protein 1 cDNA from reovirus serotype 3 strain in the pST3-S120 was kindly provided by Dr Wolfgang K Jolik from Duke University Medical Center. For its expression in E. coli, using PCR, a 1.4 kb cDNA fragment containing the restriction endonuclease sites, EcoRI and PstI, was inserted into the polylinker site of an E. coli expression plasmid, pMAL-C2 (New England Biolabs, Beverly, MA, USA). The resultant, pMAL-C2-S1, was used to transform E. coli, strain BL21 (DE3; Novagen, Madison, WI, USA). Upon induction with IPTG, the maltose-binding protein (MBP)::protein 1 fusion protein was induced in the cytoplasm of E. coli. The clear lysate of E. coli containing the fusion protein was purified by affinity chromatography using amylose resin according to manufacturer’s directions (New England Biolabs). This MBP::protein 1 fusion protein is referred to as recombinant protein 1. Preparation of recombinant fusion protein 1-polylysine–DNA complex The recombinant protein 1 was covalently linked to poly-L-lysine (PL) according to the methods of Wagner et al.4 Protein 1 was purified and resuspended in phosphate-buffered saline (PBS), pH 7.3. To generate the dithiopyridine linker, both protein 1 and PL were each modified with succinimidyl 3-(2-pyridyldithio)propionate (SPDP; Sigma Chemical, St Louis, MO, USA). Briefly, in separate vessels, 10 mg of protein 1 in 5 ml PBS, pH 7.3, and 20 mg of PL (Sigma), with an average chain length of 270 lysine monomers, in 1 ml of 75 mM sodium acetate were each vigorously mixed to react with SPDP in 15 mM ethanolic solution for 1 h. The resulting SPDP modified protein 1 was then dialyzed against PBS, pH 7.3, and the modified PL was then dialyzed against 20 mM sodium acetate to remove unbound SPDP. To generate the mercaptopropionate linker, the resultant PL with dithiopyridine linker was further mixed with 23 mg dithiothreitol (DTT) in sodium bicarbonate solution, pH 7.5, for 1 h under argon. The mercaptopropionate PL was dialyzed against 20 mM sodium acetate to remove free DTT. The 10 mg of dithiopyridine-modified protein 1 was then mixed with the 20 mg of mercaptopropionate-modified PL under argon at room temperature for 18 h. The resultant reaction generated what is referred to as protein 1-PL conjugate. This conjugate was dialyzed to remove unreacted mercaptopropionate-PL using a membrane with an exclusion of 100 kDa, against HEPES buffered saline (20 mM HEPES, 100 mM sodium chloride, pH 7.4; HS). Protein 1-PL concentration was determined using a Bradford assay (Pierce, Rockford, IL, USA). For control transfections, MBP-PL conjugates were similarly generated. For the formation of conjugate–DNA complex, the protein 1-PL conjugate in 125 l of HS was added dropwise into an equal volume of HS containing the plasmid DNA and incubated at room temperature for 30 min to form conjugate–DNA complex. Cell ligand binding assay To assess the cell-binding capacity of the protein 1 and protein 1-PL conjugates, an immunofluorescent binding assay was performed. The protein 1 and 1-PL conjugates were incubated with mouse L cells (CCL-1, American Type Culture Collection, Manassas, VA, USA), RFL-6 fibroblast cells (CCL-192, ATCC), and Caco-2 cells (HTB-37, ATCC) and binding was assessed using 20 g/ml of biotinylated monoclonal anti-reovirus protein 1 antibody (HB-167, ATCC) and SA-PE (Southern Biotech, Birmingham, AL, USA), and specific binding was then assessed using flow cytometry. No staining was obtained with biotinylated normal mouse IgG or in the presence of SA-PE only. Cell culture and transfection with plasmid DNA The mouse L cells, RFL-6 cells, and Caco-2 cells were used for targeting gene transfer by protein 1-PL conjugate. The mouse L cells have been used as the in vitro model for reovirus protein 1 binding studies. Cells were maintained in complete media: Dulbecco’s minimum essential medium (DMEM; BioWhittaker, Walkersville, MD, USA), supplemented with 10% fetal bovine serum (Life Technologies, Grand Island, NY, USA) at 37°C under 5% CO2. For Luc assay, 2.5  105 cells were added to each well of the 12-well plate and allowed to adhere overnight. The conjugate–DNA complexes were added and incubated for another 24 h in complete media. For chloroquine treatment, the cells were incubated with protein 1-PL–DNA complexes and 100 M chloroquine for 4 h at 37°C. Four hours after incubation, the conjugate–DNA complexes were removed, and cells were incubated with complete media for another 24 h. The cells were lysed to assay reporter gene activity. For -gal assay, 5 105 cells were added to each well of a six-well plate and allowed to adhere overnight. The conjugate–DNA complexes containing 8 g 1-PL and pCMV-gal (Life Technologies), with or without chloroquine, were added and incubated for 24 h. The cells were then incubated with fresh media for another 24 h before flow cytometry analysis. 105 cells were added to each well of the 12-well plate and allowed to adhere overnight. The conjugate–DNA complexes were added and incubated for another 24 h in complete media. For chloroquine treatment, the cells were incubated with protein 1-PL–DNA complexes and 100 M chloroquine for 4 h at 37°C. Four hours after incubation, the conjugate–DNA complexes were removed, and cells were incubated with complete media for another 24 h. The cells were lysed to assay reporter gene activity. For -gal assay, 5 105 cells were added to each well of a six-well plate and allowed to adhere overnight. The conjugate–DNA complexes containing 8 g 1-PL and pCMV-gal (Life Technologies), with or without chloroquine, were added and incubated for 24 h. The cells were then incubated with fresh media for another 24 h before flow cytometry analysis. Assays for reporter gene detection The Luc gene was used as a reporter gene to assay protein 1-PL conjugate-mediated transfection. A 1.4 kb Luc gene fragment flanked with HindIII and EcoRV was extracted from pSPKuci(+) (Promega, Madison, WI, USA). The pCMVLuciferase (pCMVLuc) was constructed by ligating the 1.4 kb luciferase gene into the polylinker site in pcDNA3.1(+) (Invitrogen, Carlsbad, CA, USA). The cells were lysed with 1 luciferase lysis buffer (Promega). Twenty l of supernatant of cell lysates were mixed with 100 l of Luc assay buffer and assayed with a luminometer (LUMAT LB 9507, EG&G Berthold, Germany). The relative light units from the total lysates were used to express the Luc activities produced from each transfection. Expression of -gal was visualized by incubating the transfected cells with PBS solution containing 1 mg/ml of 5-bromo-4-chloro-3-indolyl--galactopyranoside (X-gal, Boeringer Mannheim, Indianapolis, IN, USA) at 37°C for 16 h. To quantify the transfection efficiency, cells having been transfected with the constructs pCMV--Gal (Life Technologies) were harvested, loaded with 200 M fluorescein-mono-–D-galactopyranoside (FDG; Molecular Probe, Eugene, OR, USA) for 30 min at 37°C and diluted with cold PBS to a final concentration of 2.5 105 cells/ml. Flow cytometry analysis was performed using a Becton Dickinson FACSCalibur. Histochemical determination of fusion protein 1 binding to NALT NALT tissues were collected as previously described.48,49 Palates with visible NALT were washed in DMEM, and before binding with biotinylated protein 1 (following standard procedures), NALT were first incubated in DMEM alone or in the presence of 500 g/ml of protein 1 in DMEM with gentle rotation on a GeneMate orbital Shaker (Intermountain Scientific, Bountiful, UT, USA) for 45 min at 4°C. NALT were incubated with excess unmodified protein 1 in order to inhibit biotinylated protein 1 binding, and thus, show specificity of binding to the NALT. NALT were then washed gently in DMEM and incubated in 50 g/ml biotinylated protein 1 in DMEM, and were again rotated gently for 45 min at 4°C. Following incubation, NALT were removed, rinsed gently in PBS, and then arranged in 15 mm by 15 mm Tissue Tek Cryomold (Miles, Elkhard, IN, USA) with their ventral surfaces oriented toward the bottom of the mold. The palates were then frozen in Tissue Tek OCT compound embedding media and stored at -80°C until use. For immunoperoxidase staining, frozen NALT sections, previously treated with biotinylated protein 1, were cut at 5 mm, air dried, fixed in acetone at 4°C, and air dried before rehydration. Frozen sections were rehydrated in Dulbecco’s PBS (DPBS) containing 0.2% normal goat serum (NGS). A 1:250 dilution of SA-HRP conjugate (BioSource International, Camarillo, CA, USA) was added to the sections for 45 min at room temperature. The location of the HRP was visualized upon reaction with the precipitable substrate, 3-amino-ethylcarbazole (AEC; Sigma). Statistical analysis Statistical differences between experimental parameters were determined to be significant using the Student’s t test. Acknowledgements
We thank Mr Peter Hillemeyer for his assistance with flow cytometry. This work was supported by US Public Health Service Grants AI-42673, S10 RR11877, and in part by Montana Agricultural Station.
|
 1. Highly efficient uptake and internalization of protein
1. Highly efficient uptake and internalization of protein 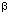 -galactosidase is visualized as dark spots by X-gal staining after transfection with protein 1-PL-pCMV
-galactosidase is visualized as dark spots by X-gal staining after transfection with protein 1-PL-pCMV g of fusion protein 1-PL and 2
g of fusion protein 1-PL and 2