| Introduction The use of adenovirus as a vector for gene delivery is becoming more and more widespread. Beside the potential of adenovirus for gene therapy, it is an efficient tool to study in vitro and in vivo gene expression in cell lines or tissues that are refractory to other gene delivery methods (for review, see Ref. 1). Despite recent progress to generate adenoviral vectors deleted for all viral genes,2,3 E1-substituted adenoviruses are still largely used. Many techniques are now available to construct such viruses. Most of them require recombination between a plasmid carrying both the left end of the adenoviral DNA and the gene of interest, and the right end of the adenoviral genome, either as a linear4 or circular5 DNA. However, these methods are time-consuming. Since the viral progeny is usually contaminated with the parental virus, at least two rounds of plaque assays are required to obtain a pure virus preparation. Screening for the recombinant virus has been facilitated by using counter-selection methods,6,7,8 by extensively fragmenting the viral DNA complexed with the adenoviral terminal protein9 or by using Cre-lox-mediated recombination.3 Alternative approaches have been developed where the sequence of the recombinant adenovirus is reconstituted either in yeast10 or in E. coli11,12,13,14,15,16 before being transfected into 293 cells.17 Such methods have the advantage that copies of the recombinant viral DNA are purified from clones and should therefore generate homogenous virus preparations. Bacterial plasmids or cosmids are even more attractive, since they are easily prepared in large quantities for transfections. Such plasmids are constructed either by recombination in E. coli11,13,14 or by in vitro ligation.15,16 The former methods11,13,14 require unfortunately multiple steps in E. coli, while the latter techniques15,16 generate the recombinant plasmid inefficiently. The only method using cosmid technology generates virus poorly.12 In this paper we describe a quick and efficient method to generate adenoviral vectors that contain expression cassettes in early region 1 (E1) or in both early regions 1 and 3 (E1 and E3). Two or four cloning steps in E. coli, respectively, and a transfection of the resulting cosmid into E1-expressing cells are sufficient to recover the recombinant virus. Cosmid construction and purification are occasionally difficult. Therefore we use positive selection methods to facilitate the construction of the recombinant cosmids. Furthermore, employing cosmids alleviates the need for extensive and time-consuming virus purification. ResultsStrategy for the construction of E1-substituted adenoviruses Our strategy to generate E1-substituted adenoviruses is based on the construction of a cosmid vector containing the sequence of the desired recombinant adenovirus. The advantage of using cosmid technology is that only DNAs with sizes ranging between 39 and 50 kb can be packaged into particles.18 Therefore this technique will facilitate the transformation of E. coli with large plasmids and counter-select clones carrying small vectors that are often generated when large DNA molecules are introduced into E. coli using conventional methods.15,16 In order to facilitate cosmid construction and analysis, the gene of interest is linked to two positive-selection markers: the cos site, necessary for DNA packaging into particles, and the sequences coding for the lacZ  -peptide. The presence of the cos site next to the gene of interest rather than in the adenoviral plasmid guarantees the presence of the gene of interest in the resulting cosmid. The juxtaposition of the lacZ promoter/operator region next to the -peptide coding region ensures the correct orientation of the insert relative to the adenoviral sequences, by a simple blue/white colony screening. -peptide. The presence of the cos site next to the gene of interest rather than in the adenoviral plasmid guarantees the presence of the gene of interest in the resulting cosmid. The juxtaposition of the lacZ promoter/operator region next to the -peptide coding region ensures the correct orientation of the insert relative to the adenoviral sequences, by a simple blue/white colony screening. The strategy is described in Figure 1. The first step is the insertion of the expression cassette into the multiple cloning sites of pAd063. This 3.2 kb intermediate vector contains the first 353 nucleotides of Ad5 DNA immediately preceded by PacI and SwaI sites, a cos site and the sequence coding for the lacZ -peptide. These sequences are enclosed between two repeats of ClaI, BstBI and Psp1406I restriction sites, which generate in-frame compatible cohesive ends. The intermediate plasmid containing the gene of interest is digested with ClaI, BstBI or Psp1406I, whichever is not present in the expression cassette, and the DNA ends are dephosphorylated using an alkaline phosphatase. The fragment containing the cos site, the adenoviral sequences (map units 0–1) and the expression cassette is purified on agarose gel. This fragment is ligated with either pAd242 or pAd244, both of them linearized with ClaI. pAd242 is a 40.5 kb plasmid that contains both the remainder of the adenoviral sequences (mu 9.2–100), with PacI and SwaI sites flanking the right inverted terminal repeat (ITR), and the lacZ promoter/operator sequences. pAd244 is identical to pAd242, except for a 2.6 kb deletion in the E3 region. The DNA is packaged in vitro into phage and then infected into E. coli. Because of the juxtaposition of the lacZ promoter/operator sequences with the sequences coding for the lacZ -peptide, colonies which contain the cosmid with the insert in the correct orientation will stain blue in the presence of X-gal. DNA is purified, digested with either PacI or SwaI to generate a linear adenoviral sequence, and finally transfected into 293 or 911 cells.17,19 Depending on the virus, the cells and the efficiency of transfection, plaques appear on average after 1 week but can be visible as early as 4 days after transfection. When properly organized, generation of a recombinant adenovirus takes only 12 days, from the first cloning step until plaques can be harvested. pAd242 and pAd244 have maximal cloning capacities of 5.0 kb and 7.7 kb, respectively. Construction of an adenovirus expressing E. coli 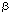 -galactosidase in the E1 region -galactosidase in the E1 region In order to demonstrate the efficiency of our method, a cassette expressing an E. coli 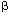 -galactosidase under the control of a human cytomegalovirus (CMV) immediate–early gene promoter and a protamine polyadenylation signal was introduced into pAd063 (Figure 2a). A BstBI fragment containing the reporter gene was purified and ligated to ClaI-digested pAd242. DNA was packaged into particles. E. coli Top10 strain (Invitrogen, Carlsbad, CA, USA) was infected and transformants were selected on Luria-Bertani (LB) medium supplemented with ampicillin, IPTG and X-gal. 193 blue colonies and 179 white colonies were obtained. Cosmid DNA was prepared from 91 blue colonies and digested with HindIII. All clones but one showed an identical restriction pattern corresponding to the insertion of the BstBI fragment in pAd242, in the correct orientation relative to the adenoviral sequence (data not shown). -galactosidase under the control of a human cytomegalovirus (CMV) immediate–early gene promoter and a protamine polyadenylation signal was introduced into pAd063 (Figure 2a). A BstBI fragment containing the reporter gene was purified and ligated to ClaI-digested pAd242. DNA was packaged into particles. E. coli Top10 strain (Invitrogen, Carlsbad, CA, USA) was infected and transformants were selected on Luria-Bertani (LB) medium supplemented with ampicillin, IPTG and X-gal. 193 blue colonies and 179 white colonies were obtained. Cosmid DNA was prepared from 91 blue colonies and digested with HindIII. All clones but one showed an identical restriction pattern corresponding to the insertion of the BstBI fragment in pAd242, in the correct orientation relative to the adenoviral sequence (data not shown). Six pAd242-Gal cosmids were purified, digested with PacI and transfected into 911 cells. On average, 12 plaques per 6 cm-diameter dish or 5 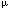 g DNA were recovered. A total of 70 plaques were isolated. Virus was amplified and used to infect A549 cells. Cells infected with the various viral clones turned blue after X-gal staining, unlike non-infected cells (Figure 2c). Viral DNA was isolated from 10 clones and digested with HindIII. No difference was observed between the original cosmid DNA and the viral progeny, indicating that no gross rearrangement had occurred upon transfection (Figure 2b). g DNA were recovered. A total of 70 plaques were isolated. Virus was amplified and used to infect A549 cells. Cells infected with the various viral clones turned blue after X-gal staining, unlike non-infected cells (Figure 2c). Viral DNA was isolated from 10 clones and digested with HindIII. No difference was observed between the original cosmid DNA and the viral progeny, indicating that no gross rearrangement had occurred upon transfection (Figure 2b). Strategy for the construction of E1- and E3-substituted adenoviruses Our system enables the construction of recombinant adenoviruses that contain two expression cassettes, one in the E1 region and the other in the E3 region, and totaling maximum 7.5 kb. As for the generation of E1- substituted viruses, cosmid technology is used. This time, each expression cassette is linked to one positive-selection marker, either the sequences coding for the lacZ -peptide, or a cos site. The insertions are performed sequentially, using two intermediate vectors (Figure 3). First, the expression cassette to be inserted into the E3 region is cloned into the multiple cloning site of pAd083. This vector contains a 173 bp-long cos site, corresponding to the minimal region essential for DNA packaging into phage .20 The resulting plasmid is digested with ClaI, BstBI or Psp1406I, whichever is not present in the gene of interest. The DNA ends are dephosphorylated, the fragment containing the expression cassette and the cos site is purified on agarose gel and cloned into the BstBI site of pAd244. Because the cos site is linked to the gene of interest, any ampicillin-resistant clone will contain a cosmid with the expression cassette inserted into the E3 region, in either orientation relative to the adenoviral sequences (intermediate cosmid). A cassette expressing the second gene of interest is inserted into another intermediate vector, pAd060. This plasmid contains the first 353 nt of Ad5 DNA flanked with PacI and SwaI sites and the sequences coding for the lacZ -peptide. The resulting plasmid is digested with ClaI, BstBI or Psp1406I, whichever is not present in the expression cassette. The DNA ends are dephosphorylated using an alkaline phosphatase and the fragment containing the sequences encoding the lacZ -peptide, the adenoviral sequences and the expression cassette is purified on agarose gel. This fragment is ligated with the intermediate cosmid linearized with ClaI. The DNA is packaged in vitro into phage and infected into E. coli. Because of the juxtaposition of the lacZ promoter/operator sequences with the sequences coding for the lacZ -peptide, colonies which contain the cosmid with the insert in the correct orientation will stain blue in the presence of X-gal. The resulting cosmid is purified, digested with either PacI or SwaI to generate a linear adenoviral sequence, and finally transfected into 293 or 911 cells. Viral plaques appear on average 7 days after transfection. Construction of an adenovirus expressing Renilla and firefly luciferases In order to illustrate the efficiency of our system, a virus expressing both Renilla and firefly luciferases in the E1 and E3 regions respectively, was constructed (Figure 4a). First, a cassette expressing a firefly luciferase under the control of an SV40 promoter and polyadenylation signal was introduced into pAd083. A ClaI fragment containing the reporter gene was purified and ligated to BstBI-digested pAd244. DNA was packaged into particles and infected into E. coli. Cosmids from six ampicillin-resistant clones were characterized by restriction analysis. All contained the firefly luciferase expression cassette, in either orientation (data not shown). The resulting cosmid, pAd244-FL, was then used in the second step of the procedure. A cassette expressing a Renilla luciferase under the control of an SV40 promoter and polyadenylation signal was cloned into pAd060. The resulting plasmid was digested with ClaI and the fragment containing the sequences encoding the Renilla luciferase was purified and ligated with ClaI-digested pAd244-FL. After DNA packaging into phage and E. coli infection, 135 blue colonies and 429 white colonies were obtained. Cosmids were purified from five of the blue colonies and revealed, through restriction analysis, an insert in the correct orientation relative to the adenoviral sequences (data not shown). One cosmid was further purified and digested with PacI. From two transfection experiments into 911 cells, 10 plaques were recovered, giving an average of five plaques per 6 cm-diameter dish or 5 g DNA. Virus was expanded and used to infect A549 cells. After 2 days of expression, cells were lyzed and a dual luciferase assay was performed (Figure 4c). All viral clones appeared to express both luciferases. The presence of both expression cassettes in the viral genome was confirmed by restriction analysis of the viral DNAs (Figure 4b). Next, we wanted to investigate whether the presence of an expression cassette in the E3 region has an effect on viral infectivity and titer. We constructed another virus, AdM15, which expresses the -galactosidase  M15 mutant under the control of a CMV promoter and the lacZ -peptide under the control of a RSV promoter, respectively, in the E1 and E3 regions. As a control, we constructed an adenovirus, AdM15, expressing the -galactosidase M15 mutant under the control of a CMV promoter, in the E1 region, leaving the E3 region empty. From a single transfection experiment in 911 cells, cosmids pAdM15 and pAdM15 yielded eight and one plaques, respectively. For both viruses, the first plaques appeared 7 days after transfection. We prepared crude viral extracts from 293 cells infected with each of these viruses and determined the viral titers by plaque assays. It appeared that AdM15 grew faster and yielded a seven-fold higher titer than AdM15 (6 M15 mutant under the control of a CMV promoter and the lacZ -peptide under the control of a RSV promoter, respectively, in the E1 and E3 regions. As a control, we constructed an adenovirus, AdM15, expressing the -galactosidase M15 mutant under the control of a CMV promoter, in the E1 region, leaving the E3 region empty. From a single transfection experiment in 911 cells, cosmids pAdM15 and pAdM15 yielded eight and one plaques, respectively. For both viruses, the first plaques appeared 7 days after transfection. We prepared crude viral extracts from 293 cells infected with each of these viruses and determined the viral titers by plaque assays. It appeared that AdM15 grew faster and yielded a seven-fold higher titer than AdM15 (6 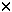 1011 p.f.u./ml versus 9 1010 p.f.u./ml, in triplicate experiments). These data show that the insertion of an expression cassette in the E3 region can have an effect on virus replication. 1011 p.f.u./ml versus 9 1010 p.f.u./ml, in triplicate experiments). These data show that the insertion of an expression cassette in the E3 region can have an effect on virus replication. DNA yield and cosmid stability Because large plasmids are frequently unstable in E. coli and difficult to purify, we investigated the stability and yield of the adenoviral cosmids. To assess the stability of our vectors, bacteria containing cosmid pAd242-Gal were grown in liquid LB medium at 37°C. The culture was diluted 1000-fold each time OD600 2.0. After 48 h, the bacterial culture was purified on an agar plate. Ten clones were grown and revealed to contain the original cosmid as judged by restriction analysis. However, when the bacteria were cultured extensively in the stationary phase, rearrangements were observed (data not shown). Since the harvest time seemed to be important, we also analyzed the dependence of cosmid DNA yield on bacterial cell density. Bacteria containing cosmid pAd242-Gal were grown in liquid LB at 37°C. Culture aliquots were taken every 30 min, cosmid DNA was purified, digested with HindIII, and analyzed on agarose gel. DNA amounts were estimated by densitometry, after ethidium bromide staining. Figure 5 shows that cosmid yield is strongly dependent on the bacterial growth phase: it is maximal at the end of the exponential phase and drops dramatically as the cells enter the stationary phase. Taken together, our data suggest that cosmid DNA purification should be performed using freshly grown bacteria, harvested at the end of the exponential growth phase. DiscussionThe past few years have seen the proliferation of methods for the generation of recombinant adenoviral vectors. The method we propose in this paper has the following advantages. First, like several other methods,11,12,13,14 the reconstitution of the sequence of the desired recombinant adenovirus in E. coli facilitates virus purification, since the transfection is performed using DNA purified from a single clone. One agar overlay on the transfected cells should therefore be sufficient to isolate a homogenous virus preparation. Second, as has already been suggested,12 cosmid technology is particularly well suited for the cloning of the 36 kb-long adenoviral genome. Because of its requirement for large DNAs, the method selects clones containing full-size genomes, and excludes clones carrying small recombination products that are often generated when large plasmids are introduced into E. coli using conventional transformation methods like heat shock or electroporation.15,16 In our method, we use two positive- selection markers (a cos site and the lacZ sequences) to facilitate the generation of the correct cosmid. The whole procedure is very efficient, since a few hundred cosmid clones are usually recovered. Third, our method does not require any homologous recombination event, either in E. coli,11,13,14 yeast,10 or mammalian cells.4,21 In mammalian cells, the recombination step is probably rate-limiting, but the advantage is that only the correct product of recombination should generate virus. In contrast, constructing the adenoviral genome by recombination in E. coli or yeast represents a major drawback since no selection is available to guarantee an infectious DNA. Unpredicted recombination events can occur, especially in yeast (X Danthinne, unpublished results), and without a thorough analysis of the recombinant plasmid or YAC, clones can be selected that are unable to generate virus. Furthermore, recombination in E. coli requires the use of a recA+ strain, such as BJ5183.11,14 The transfer of the adenoviral plasmid, product of recombination, into a recA endA strain is necessary to obtain high DNA yields for transfection. This additional step is not required when using the cosmid approach, since the infection of E. coli with bacteriophage is easily performed using recA endA strains such as DH5, XL1-blue (Stratagene, La Jolla, CA, USA) or Top10 (Invitrogen, Carlsbad, CA, USA). Thus, using ligation to construct the genome of the recombinant virus in E. coli has strong advantages. Fourth, the proposed method allows the construction of double recombinant adenoviruses, containing one expression cassette in the E1 region and the other in the E3 region. With three cloning steps in E. coli and a transfection, our strategy is maybe no faster than other existing methods,21 but probably easier since each expression cassette is inserted into the cosmid together with a positive selectable marker (either a minimal cos site, or the sequences encoding the lacZ -peptide). So far, viruses expressing two transgenes from independent promoters have not been used extensively,22,23,24 probably because of the difficulties in constructing them. Yet they are invaluable tools in case one needs to express a protein composed of two different subunits or two proteins acting synergistically. The method we have described in this paper should facilitate the construction of such viruses. Fifth, the method described in this paper is very versatile as far as the choice of restriction enzymes for cloning is concerned. Indeed, unlike all other existing methods, we have flanked both adenoviral ITRs with two rare- cutting restriction enzymes (PacI and SwaI), to linearize the cosmid DNA before transfection. Three enzymes (ClaI, BstBI and Psp1406I) are also available to transfer the expression cassettes from the intermediate plasmids (pAd063, pAd083 and pAd060) to the adenoviral plasmids (pAd242, pAd244). It is therefore likely that this method will be useful in a high number of applications. Using our vectors, the construction of an E1-substituted adenovirus will not be possible if the expression cassette contains simultaneously a ClaI, a BstBI and a Psp1406I site, or simultaneously a SwaI and a PacI site. The construction of an E1- and E3-substituted adenovirus will not be possible if the expression cassette to be inserted into the E3 region contains a ClaI site, or if the expression cassette to be inserted into the E1 region contains simultaneously a ClaI, a BstBI and a Psp1406I site, or if both SwaI and PacI sites are present in either or both expression cassettes. Finally, the method proposed in this paper is fast since E1-substituted viruses can be obtained in as little as 12 days, which is comparable to other methods currently available. The method is very reliable: in both above-described examples, 90 out of 91 cosmid clones were found correct by restriction analysis, all seven cosmids that were transfected into 911 cells were infectious and all plaques that were generated expressed the transgene(s). A first limitation of the method is the possibility of cosmid recombination in E. coli, simultaneously with a decrease of DNA yield. As shown in Figure 5, these potential hurdles can be avoided if the bacteria are harvested before the cells enter the stationary growth phase. Also, we have observed that the nature of the transgene inserted in the E1 region (pAd063-pAd242) has a slight effect on the intensity of the blue colonies, probably because of transcriptional interference between the transgene and the lacZ transcription units in E. coli (not shown). Another limitation of the technique is the size of the expression cassette that can be introduced in the adenoviral genome. Our vectors allow the insertion of a maximum of 7.7 kb in the E1 region alone, or a total of 7.5 kb in both the E1 and E3 regions, due to the fact that the expression cassette inserted in the E3 region is flanked by a cos site. Other techniques, now available, allow the insertion of larger DNA fragments. For instance, up to 10 kb of transgene sequences can be introduced in the E1 region using a set of vectors deleted for the E4 region and an E4-complementing cell line.14 Also, a total of 8.1 kb of transgene sequences can be introduced into both the E1 and E3 region, using vectors which contain expanded deletions in the E1 and E3 regions.21 In conclusion, we have proposed in this paper a quick and efficient method to generate recombinant adenoviral vectors. Importantly, the method does not require any recombination event and allows the insertion of two expression cassettes, in the E1 and E3 regions of the adenoviral genome. It should therefore be useful for a large number of applications. Materials and methodsPlasmid constructions The plasmids described in this paper were constructed using PCR-based methods and many intermediate steps that are too long to be described here. The authors will provide a detailed description upon request and will be delighted to share their vectors and start up new collaborations with other researchers. pAd060 (Figure 3) was constructed by replacing the ampicillin-resistance gene of pUC19 with a kanamycin-resistance gene isolated from Tn903.25 The sequences encoding the lacZ -peptide, the first 353 nucleotides from the Ad5 genome (including the left ITR and the adenoviral packaging signal), and a multiple cloning site were inserted between a tandem repeat of ClaI, BstBI and Psp1406I sites. pAd063 (Figure 1) was constructed by inserting a cos site between the lacZ and the adenoviral sequences of pAd060. pAd083 (Figure 3) was made by deleting the lacZ sequences from pAd063. To construct pAd242 (Figure 1), the lacZ -peptide sequences of pUC19 were replaced by the right end of Ad5 genome (nt 3300-end) and a 5.7 kb DNA fragment from phage . pAd244 (Figures 1 and 3) is a similar to pAd242, except that the E3 sequences corresponding to nt 28133–30818 in the Ad5 genome have been deleted and replaced with a unique BstBI site. Cosmid constructions and purification Packaging of DNA into phage was performed using Gigapack III extract according to the manufacturer (Stratagene). Cosmid DNA was prepared from fresh bacteria grown in LB and harvested when OD600 2.0, ie before reaching the stationary phase. DNA was prepared by the alkaline lysis method,26 and further purified on a CsCl-ethidium bromide gradient or using purification columns such as StrataPrep EF (Stratagene), Nucleobond (Clontech, San Francisco, CA, USA), or Wizard Purefection (Promega, Madison, WI, USA). Virus generation and analysis Cosmid DNA was linearized with PacI or SwaI, ethanol-precipitated and resuspended in sterile TE pH 7.5. DNA transfection into 911 or 293 cells, virus purification, and viral DNA characterization were performed as previously described.27 Reporter gene vectors and assays A 4.3 kb cassette expressing E. coli -galactosidase was purified by digesting plasmid pNCMVlacF (Dr RD Gerard, Center for Transgene Technology and Gene Therapy, Leuven, Belgium) with SpeI and BglII. -Galactosidase assays were performed on A549 cell monolayers (ATCC CCL185) as previously described.28 Cassettes expressing firefly and Renilla luciferases were obtained, respectively, from plasmids pGL3 (2.2 kb ClaI fragment) and pRLSV40 (1.8 kb BglII–BamHI fragment) (Promega). Dual luciferase assays were performed according to the protocol provided by Promega. DNA analyses DNA quantifications by densitometry were carried out using an Alpha Imager 2000 (Alpha Innotech, San Leandro, CA, USA). DNA sequence analyses were performed using the GCG software package (Madison, WI, USA), through the Belgian EMB Node facility. Acknowledgements
We gratefully acknowledge Drs E Chang, Y Laroche and D Salmi for critical review of the manuscript, and Dr RE Vestal for generous financial help.
|
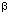 -galactosidase Proc Natl Acad Sci USA 1996 93: 5731–5736 MEDLINE
-galactosidase Proc Natl Acad Sci USA 1996 93: 5731–5736 MEDLINE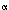 : promoter/operator region and
: promoter/operator region and  g/ml ampicillin and aliquots were harvested every 30 min. Cosmid DNA was purified and quantified by densitometry after HindIII digest and agarose gel electrophoresis.
g/ml ampicillin and aliquots were harvested every 30 min. Cosmid DNA was purified and quantified by densitometry after HindIII digest and agarose gel electrophoresis.